
Research activity

General method for solving Schrödinger and Dirac equations
Schrödinger equation (SE) is the most fundamental equation in chemistry. If its exact solution can be obtained, we can accurately predict all chemical phenomena. Therefore, solving the SE has invaluable significance in both pure and applied sciences. Recently, Nakatsuji has realized this dream: a general method for solving the analytical solution of the SE has been developed. In addition, Nakatsuji proposed Scaled Schrödinger equation which is equivalent to the SE. With these breakthroughs, we succeeded first in the world to solve analytical solution of the SE for many-electron systems, which was the long-cherished dream of theoretical chemist. Since our method is based on general theory, it is applicable to large systems. Our recent calculations established new world records regarding the precision of the solutions in many systems so far applied.
In our recent study, we extended our method to calculate heavy elements in which the relativistic effect become important. Using the Dirac-Coulomb equation (DCE) as the fundamental equation, we re-formulated our method and succeeded to obtain the analytical solution of DCE first in the world. In addition, since our method is applicable to the time-dependent SE, we can also study electron dynamics with very high accuracy.
This method has a great possibility for bringing a paradigm shift in quantum chemistry, and its future development is highly appreciated.
- Scaled Schrödinger Equation and the Exact Wave Function, H. Nakatsuji, Phys. Rev. Lett. 93, 030403 (2004).
- General Method of Analytically Solving the Schrödinger Equation of Atoms and Molecules, H. Nakatsuji, Phys. Rev. A, 72, 062110 (2005).
- Deepening and Extending the Quantum Principles in Chemistry, H. Nakatsuji, Bull. Chem. Soc. Jpn. 78, 1705 (2005).
- Analytically Solving the Dirac-Coulomb Equation for Atoms and Molecules, H. Nakatsuji H. Nakashima, Phys. Rev. Lett., 95, 050407 (2005).
- Free ICI (Iterative Complements Interaction) Calculations of Hydrogen Molecule, Y. Kurokawa, H. Nakashima, and H. Nakatsuji, Phys. Rev. A, 72, 062502 (2005).
- Structure of the Exact Wave Function, H. Nakatsuji, J. Chem. Phys., 113, 2949-2956 (2000).
- Structure of the Exact Wave Function. II. Iterative Configuration Interaction Method, H. Nakatsuji and E. R. Davidson, J. Chem. Phys. 115, 2000-2006 (2001).
- Structure of the Exact Wave Function. III. Exponential Ansatz, H. Nakatsuji, J. Chem. Phys., 115, 2465-2475 (2001).
- Structure of the Exact Wave Function. IV. Excited Sates from Exponential Ansatz and Comparative Calculations by the Iterative Configuration Interaction and Extended Coupled Cluster Theories, H. Nakatsuji, J. Chem. Phys., 116, 1811-1824 (2002).
- Structure of the Exact Wave Function. V. Iterative Configuration Interaction Method for Molecular Systems within Finite Basis, H. Nakatsuji and M. Ehara, J. Chem. Phys., 117, 9-12 (2002).
- Iterative CI General Singles and Doubles (ICIGSD) Method for Calculating the Exact Wave Functions of the Ground and Excited States of Molecules, H. Nakatsuji, M. Ehara, J. Chem. Phys. 112, 194108 (2005).
- Inverse Schrödinger Equation and the Exact Wave Function, H. Nakatsuji, Phys. Rev. A 65, 052122 (2002).
- Deepening and Realization of the Quantum Principles in Chemistry, H. Nakatsuji, 40th IUPAC Congress, Beijing, China, August 14-19 (2005)
Theory for studying chemistry of excited states: SAC-CI method

The SAC/SAC-CI method is an accurate electronic-structure theory and applicable to study wide-range of molecular sciences. The SAC/SAC-CI method was proposed by Nakatsuji in 1978. Since then, the method has been recognized as the most advanced method.
The function of the SAC-CI program published in Gaussian03 package is summarized in the figure. The SAC-CI program is applicable to wide-range of chemistry. We are continuously developing new functions and algorithms, which will also be released in the forthcoming version of Gaussian. Our method would manifest more and more important contributions in the field of the excited-states chemical reaction and fine theoretical spectroscopy.
- SAC-CI website: http://www.sbchem.kyoto-u.ac.jp/nakatsuji-lab/sacci.html
- Cluster Expansion of the Wavefunction. Pseudo-Orbital Theory Applied to Spin Correlation, H. Nakatsuji and K. Hirao, Chem. Phys. Lett., 47(3), 569-571 (1977).
- Cluster Expansion of the Wavefunction. Symmetry-Adapted-Cluster (SAC) Expansion, Its Variational Determination, and Extension of Open-Shell Orbital Theory, H. Nakatsuji and K. Hirao, J. Chem. Phys., 68(5), 2053-2065 (1978).
- Cluster Expansion of the Wavefunction. Excited States, H. Nakatsuji, Chem. Phys. Lett., 59(2), 362-364 (1978).
- Cluster Expansion of the Wavefunction. Electron Correlations in Ground and Excited States by SAC (Symmetry-Adapted-Cluster) and SAC-CI Theories, H. Nakatsuji, Chem. Phys. Lett., 67(2,3), 329-333 (1979).
- Cluster Expansion of the Wavefunction. Calculation of Electron Correlations in Ground and Excited States by SAC and SAC-CI Theories, H. Nakatsuji, Chem. Phys. Lett., 67(2,3), 334-342 (1979).
- Electronic Structures of Ground, Excited, Ionized, and Anion States Studied by the SAC/SAC-CI Theory, H. Nakatsuji, Acta Chimica Hungarica, Models in Chemistry, 129(5), pp.719-776 (1992).
- SAC-CI Method: Theoretical Aspects and Some Recent Topics, H. Nakatsuji, in Computational Chemistry - Reviews of Current Trends, Vol. 2, p. 62-124 (1997).
- Description of Two- and Many-Electron Processes by the SAC-CI Method, H. Nakatsuji, Chem. Phys. Lett., 177(3), 331-337 (1991).
- SAC-CI Method Applied to High-Spin Multiplicity, H. Nakatsuji and M. Ehara, J. Chem. Phys., 98(9), 7179-7184 (1993).
- SAC-CI and Full CI Calculations for the Singlet and Triplet Excited States of H2O, H. Nakatsuji, K. Hirao, and Y. Mizukami, Chem. Phys. Lett., 179(5,6), 555-558 (1991).
- Analytical Energy Gradient of the Ground, Excited, Ionized and Electron-Attached States Calculated by the SAC/SAC-CI Method, T. Nakajima and H. Nakatsuji, Chem. Phys. Lett., 280 (1,2) 79-84 (1997).
- Analytical Energy Gradients of the Excited, Ionized and Electron-Attached States Calculated by the SAC-CI General-R Method, M. Ishida, K. Toyoda, M. Ehara and H. Nakatsuji, Chem. Phys. Lett., 347, 493-498 (2001).
- Electronic Excitation Spectra of Furan and Pyrolle: Revisit by the SAC-CI Method, J. Wan, J. Meller, M. Hada, M. Ehara, and H. Nakatsuji, J. Chem. Phys., 113(18), 7853-7866 (2000).
- Inner-Shell Ionizations and Satellites Studied by the Open-Shell Reference Symmetry-Adapted Cluster/Symmetry-Adapted Cluster Configuration-Interaction Method, Y. Ohtsuka and H. Nakatsuji, J. Chem. Phys. 124, 054110-1-5 (2006).
Theoretical fine-spectroscopy
As shown in the figures, our recent study has realized significantly-fine theoretical spectroscopy which highly resemble to the experimental one. In addition, our method is able to predict the structure of molecules in their excited state. The scope of the application has been broadened to core-electron processes and metal surfaces.
Out target is now expanding to the photochemistry of the photo-functional molecules. As an example, the figure compares the experimental spectrum with the SAC-CI theoretical one including the thermal effect of the internal bond rotation.
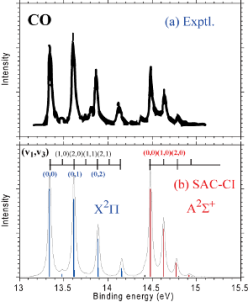
- Symmetry-Resolved Vibrational Spectra of Carbon K-shell Photoelectron Satellites in Carbon Monoxide: Experiment and Theory, K. Ueda, M. Hoshino, T. Tanaka, M. Kitajima, H. Tanaka, A. De Fanis, Y. Tamenori, M. Ehara, F. Oyagi, K. Kuramoto, H. Nakatsuji, Phys. Rev. Lett., 94, 243004 1-4 (2005).
- C1s and O1s photoelectron satellite spectra of CO with the symmetry-dependent vibrational excitations, M. Ehara, K. Kuramoto, H. Nakatsuji, M. Hoshino, T. Tanaka, M. Kitajima, H. Tanaka, Y. Tamenori, A. De Fanis, K. Ueda, J. Chem. Phys. 125, 114304-1-10 (2006).
- Electronic Circular Dichroism Spectrum of Uridine Studied by the SAC-CI method, S. Bureekaew, J. Hasegawa, H. Nakatsuji, Chem. Phys. Lett. 425, 367-371 (2006).
- Fine Theoretical Spectroscopy Using SAC-CI General-R Method: Outer- and Inner-Valence Ionization Spectra of N2O and HN3, M. Ehara, S. Yasuda and H. Nakatsuji, Z. Phys. Chem., 217, 161-176 (2003).
- Fine Theoretical Spectroscopy Using SAC-CI General-R Method: Outer- and Inner-Valance Ionization Spectra of CS2 and OCS, M. Ehara, M. Ishida, and H. Nakatsuji, J. Chem. Phys., 117, 3248-3255 (2002).
- Electronic Excitation Spectra of Furan and Pyrolle: Revisited by the SAC-CI Method, J. Wan, J. Meller, M. Hada, M. Ehara, and H. Nakatsuji, J. Chem. Phys., 113(18), 7853-7866 (2000).
- Hyperfine Splitting Constants Studied by the SAC-CI Method, H. Nakatsuji, M. Ehara, and T. Momose, J. Chem. Phys., 100(8), 5821-5828 (1994).
- Calculation of Hyperfine Splitting Constants with Slater-Type Cusp Basis by the SAC-CI Theory, H. Nakatsuji and M. Izawa, J. Chem. Phys., 91(9), 6205-6214 (1989).
Biological quantum chemistry
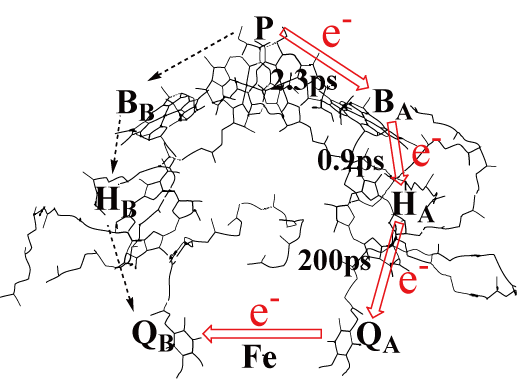
Photosynthesis, vision, and bioluminescence are one of the most intriguing research subjects, for they are life phenomenons which explicitly use the light, excited states. Photosynthesis of the green plants is very important reaction producing the energy on the globe. We have solved some enigmatic questions regarding to the photo-induced electron-transfer in the reaction center. For vision, we are studying the color-tuning mechanism of the retinal proteins. We are also studying the molecular mechanism of the bioluminescence of the fireflies and jerryfishes.
The oxygen-fixation process is essential for the living systems. We are studying the mechanism of the oxygen-molecule binding by through that observed in oxyheme and oxyhemocyanin.
- Red Light in Chemiluminescence and Yellow-green Light in Bioluminescence: Color-tuning Mechanism of Firefly, Photinus pyralis, studied by the SAC-CI method, Naoki Nakatani, Jun-ya Hasegawa, Hiroshi Nakatsuji, J. Am. Chem. Soc. in press.
- Theoretical Studies on the Color-Tuning Mechanism in Retinal Proteins, K. Fujimoto, S. Hayashi, J. Hasegawa, and H. Nakatsuji, J. Chem. Theory Comput. 3, 605-618 (2007).
- On the color-tuning mechanism of Human-Blue visual pigment: SAC-CI and QM/MM study, K. Fujimoto, J. Hasegawa, S. Hayashi, and H. Nakatsuji, Chem. Phys. Lett. 432, 252-256 (2006).
- Mechanism of Color-Tuning in Retinal Proteins: SAC-CI and QM/MM Study, K. Fujimoto, J. Hasegawa, S. Hayashi, S. Kato, H. Nakatsuji, Chem. Phys. Lett., 414, 239-242 (2005).
- On the O2 binding of Fe-porphyrin, Fe-porphycene, and Fe-corrphycene complexes, H. Nakashima, J. Hasegawa, H. Nakatsuji, J. Comp. Chem., 27, 1363-1372 (2006).
- Energetics of the Electron Transfer from Bacteriophephytin to Ubiquinone in the Photosynthetic Reaction Center of Rhosopseudomonas Viridis: Theoretical Study, J. Hasegawa, M. Ishida, and H. Nakatsuji, J. Phys. Chem. B, 107, 838-847 (2003).
- Electron Transfer in the C-Type Cytochrome Subunit of the Photosynthetic Reaction Center of Rhodopseudomonas Viridis: Ab Initio Theoretical Study, Y. Ohtsuka, K. Ohkawa, H. Nakatsuji, J. Comp. Chem., 22, 521-527 (2001).
- The Role of Proteins in the Electron Transfer in the Photosynthetic Reaction Center of Rhodopseudomonas Viridis: Bacteriopheophytin to Ubiquinone, H. Ito and H. Nakatsuji, J. Comp. Chem., 22, 265-272 (2001).
- Excited States and Electron Transfer Mechanism in the Photosynthetic Reaction Center of Rhodopseudomonas Viridis: SAC-CI Study, H. Nakatsuji, J. Hasegawa, and K. Ohkawa, Chem. Phys. Lett., 296 (5,6), 499-504 (1998).
- Excited States of the Photosynthetic Reaction Center of Rhodopseudomonas Viridis: SAC-CI Study, J. Hasegawa, K. Ohkawa, H. Nakatsuji, J. Phys. Chem. B, 102 (50), 10410-10419 (1998).
Giant SAC/SAC-CI method

We are developing SAC/SAC-CI method applicable to giant systems like molecular crystals and biological systems. Based on the view point that the entire system is the aggregate of local systems, truly large systems become our computational target.
As applications of this method, we want to clarify the excited state of the molecular crystal, the role of protein in biological reactions, and the solvation effect in chemical reactions which sometime becomes essentially important.
- Symmetry-adapted-cluster/symmetry-adapted-cluster configuration interaction methodology extended to giant molecular systems: Ring molecular crystals, H. Nakatsuji, T. Miyahara, R. Fukuda, J. Chem. Phys. 126, 084104 (2007).
Surface chemistry
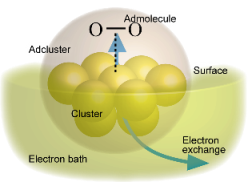
Solid catalysts have long been called as "Magic stone". Even though they are practically very important, their essential part of the catalytic mechanism has not still unveiled in many reactions. We have revealed the catalytic mechanism of various metal surfaces with our theoretical method.
Figure shows concept of the Dipped-Adcluster Model (DAM) for studying the catalytic metal surfaces. With this model, we are studying the mechanism of the catalytic metal-surfaces and photocatalytic reactions.
- Dipped Adcluster Model for Chemisorptions and Catalytic Reactions on a Metal Surface, H. Nakatsuji, J. Chem. Phys., 87(8), 4995-5001 (1987).
- Dipped Adcluster Model for Chemisorptions and Catalytic Reactions on a Metal Surface: Image Force Correction and Applications to Pd-O2 Adclusters, H. Nakatsuji, H. Nakai, and Y. Fukunishi, J. Chem. Phys., 95(1), 640-647 (1991).
- Dipped Adcluster Model for Chemisorption and Catalytic Reactions, H. Nakatsuji, Progress in Surface Science, Vol. 54, p. 1-68 (1997).
- Theoretical Study on Molecular and Dissociative Chemisorptions of an O2 Molecule on an Ag Surface: Dipped Adcluster Model Combined with SAC-CI Method, H. Nakatsuji and H. Nakai, Chem. Phys. Lett., 174(3,4), 283 (1990).
- Theoretical Studies on the Catalytic Activity of Ag Surface for the Oxidation of Olefins, H. Nakatsuji, Z. M. Hu, and H. Nakai, Intern. J. Quantum. Chem., 65, 839-855 (1997).
- Electron Transfer and Back-Transfer in the Partial Oxidation of Ethylene on an Ag Surface: Dipped Adcluster Model Study, H. Nakatsuji, K. Takahashi, and Z.M Hu, Chem. Phys. Lett., 277(5,6), 551-557 (1997).
- Activation of O2 on Cu, Ag, and Au Surfaces for the Epoxidation of Ethylene: Dipped Adcluster Model Study, H. Nakatsuji, Z. M. Hu, H. Nakai and K. Ikeda, Surf. Sci., 387 328-341 (1997).
- Mechanism of the Hydrogenation of CO2 to Methanol on a Cu(100) Surface: Dipped Adcluster Model Study, Z. M. Hu, K. Takahashi, and H. Nakatsuji, Sur. Sci., 442,(1), 90-106 (1999).
- Mechanism of Methanol Synthesis on Cu(100) and Zn/Cu(100) Surfaces: Comparative Dipped Adcluster Model Study, H. Nakatsuji and Zhen-Ming Hu, Intern. J. Quantum Chem., 77, 341-349 (2000).
- Theoretical Surface Spectroscopy of NO on the Pt(111) Surface with the DAM (Dipped Adcluster Model) and the SAC-CI Method, H. Nakatsuji, N. Matsumune, and K. Kuramoto, J. Chem. Theo. Comp. 1, 239-247 (2005).
Electronic mechanisms of NMR chemical shifts
NMR chemical shifts are very widely used in analytical chemistry but it is not well known that they involve a lot of information about the electronic structure of molecules. A purpose of the study is to clarify the electronic mechanisms of the metal chemical shifts and to offer the means for understanding the natures of bonding in the metal complexes.
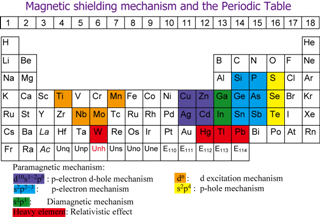
Since the chemical shift measures the angular momenta of electrons induced around the resonant nuclei by the applied magnetic field, the p- and/or d-orbital electronic structures of the metal complexes are reflected to their chemical shifts. Nakatsuji has shown that the primary mechanisms of the metal chemical shifts are the intrinsic properties of the resonant nuclei characterized by their positions in the periodic table.
The relativistic effects, spin-orbit effect in particular, are very important for the chemical shifts of molecules including heavy elements. We have developed a method to calculate these relativistic effects. We showed for the first time that the spin-orbit effect is the dominant origin of the proton and 13C chemical shifts in the HX and CH3X (X=F, Cl, Br, I) series of molecules, respectively. We performed later the Dirac-Fock four-spinor calculations of various molecules in the magnetic field. The relativistic effects and the electron correlation effects couple strongly, so that they must be calculated at the same time.
- Electronic Mechanisms of Metal Chemical Shifts from Ab Initio Theory, H. Nakatsuji, in Nuclear Magnetic Shielding and Molecular Structure Ed. by J. A. Tossell, NATO ASI Series, C386, Reidel, Dordrecht, 263-278 (1993).
- Electronic Origin of 95Mo-NMR Chemical Shift in Some Molybdenum Complexes. Relationship between Excitation Energy and Chemical Shift, H. Nakatsuji, M. Sugimoto and S. Saito, Inorg. Chem., 29(17), 3095-3097 (1990).
- Theoretical Study on Metal NMR Chemical Shifts. Gallium Compounds, M. Sugimoto, M. Kanayama and H. Nakatsuji, J. Phys. Chem., 97(22), 5868-5874 (1993).
- pin-Orbit Effect on the Magnetic Shielding Constant Using Ab Initio UHF Method, H. Nakatsuji, H. Takashima, M. Hada, Chem. Phys. Lett., 233, 95-101 (1995).
- Spin-orbit Effect on the Magnetic Shielding Constant Using the ab initio UHF Method: Tin Tetrahalides, H. Kaneko, M. Hada, T. Nakajima, and H. Nakatsuji, Chem. Phys. Lett., 261, 1-6 (1996).
- Quasi-Relativistic Theory for the Magnetic Shielding Constant. II. Gauge-Including Atomic Orbitals and Applications to Molecules, R. Fukuda, M. Hada, and H. Nakatsuji, J. Chem. Phys., 118, 1027-1035 (2003).
- Quasi-Relativistic Theory for the Magnetic Shielding Constant. III. Quasi-Relativistic Second-Order Møller Plesset Perturbation Theory and its Application to Tellurium Compounds, R. Fukuda and H. Nakatsuji, J. Chem. Phys. 123, 044101-1-10 (2005).
Development of relativistic quantum chemistry
We have long been advocating to the world about the importance of the relativistic effect in chemical phenomenon involving the heavy elements. Relativity is indispensable to modern quantum chemistry. Including both the electron-correlation effect and relativistic effect by using the Dirac equation as the fundamental equation, we are developing relativistic quantum chemistry as an ultimate theory.

- Relativistic Configuration Interaction and Coupled Cluster Methods Using Four-Component Spinors: Magnetic Shielding Constants of HX and CH3X (X = F, Cl, Br, I) M. Kato, M. Hada, R. Fukuda, H. Nakatsuji, Chem. Phys. Lett., 408, 1150-156 (2005).
- Dirac-Fock Calculations of the Magnetic Shielding Constants of Protons and Heavy Nuclei in XH2 (X=O, S, Se, and Te): A Comparison with Quasi-Relativistic Calculations, M. Hada, R. Fukuda, and H. Nakatsuji, Chem. Phys. Lett., 321, 452-458 (2000).
- Dirac-Fock Calculations of Magnetic Shielding Constants: Hydrogen Molecule and Hydrogen Halides, M. Hada, Y. Ishikawa, J. Nakatani, and H. Nakatsuji, Chem. Phys. Lett., 310 (3,4), 342-346 (1999).
- Relativistic Theory of the Magnetic Shielding Constant: A Dirac-Fock Finite Perturbation Study, Y. Ishikawa, T. Nakajima, M. Hada, and H. Nakatsuji, Chem. Phys. Lett., 283(1,2), 119-124 (1998).
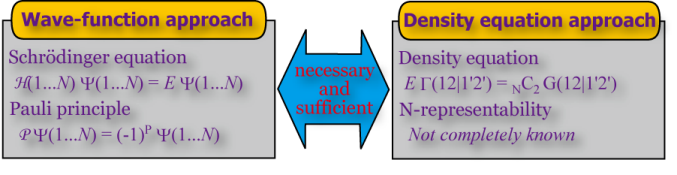
Directly solving 2nd-order density matrix without using wavefunction
Since any basic operators of properties include only one- and two-electron operators, all the properties of molecules can be calculated if the exact second-order density matrices are given. The second-order density matrix depends only on 4 electron coordinates at maximum. Therefore, we may construct quantum mechanics using the second-order density matrix as a basic variable instead of the wave function (Wave Mechanics without Wave).
Nakatsuji presented in 1976 a basic equation called density equation (or later called contracted Schrödinger equation, again in the West World) which is equivalent to the Schrödinger equation in the space of the density-matrix. In 1996, by improving Valdemoro's theory, Nakatsuji and Yasuda firstly solved the density equation for real molecules. In 2001, Nakatsuji and Nakata further invented a variational method for directly solving the second-order density matrices of molecules using positive semi-definite programming (SDP) algorithm. This method was further shown to be stable under the multi-reference and strong-correlation situations.
- Equation for the Direct Determination of the Density Matrix, H. Nakatsuji, Phys. Rev., A14, 41 (1976).
- Equation for the Direct Determination of the Density Matrix: Time- Dependent Density Equation and Perturbation Theory, H. Nakatsuji, Theor. Chem. Acc. 102, 97-104 (1999).
- Direct Determination of the Quantum-Mechanical Density Matrix Using the Density Equation, H. Nakatsuji and K. Yasuda, Phys. Rev. Lett., 76, 1039-1042 (1996).
- Direct Determination of the Quantum-Mechanical Density Matrix Using the Density Equation. II., K. Yasuda and H. Nakatsuji, Phys. Rev. A 56, 2648-2657 (1997).
- Density Equation Theory in Chemical Physics, H. Nakatsuji, in Many-electron Densities and Reduced Density Matrices, edited by J. Cioslowski, Kluwer Academic, New York 2000, pp85-116.
- Variational Calculations of Fermion Second-Order Reduced Density Matrices by Semi- definite Programming Algorithm, M. Nakata, H. Nakatsuji, M. Ehara, M. Fukuda, K. Nakata, and K. Fujisawa, J. Chem. Phys., 114, 8282-8292 (2001).
- Density Matrix Varitional Theory: Application to the Potential Energy Surfaces and Strongly Correlated Systems, M. Nakata, M. Ehara, and H. Nakatsuji, J. Chem. Phys., 116, 5432-5439 (2002).
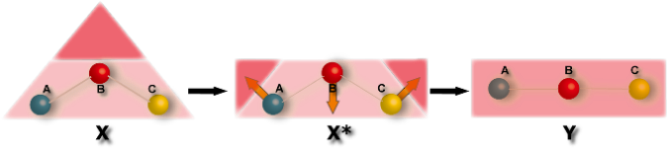
Force concept in chemistry
Nakatsuji proposed a conceptual force model, called ESF (electrostatic force) model for molecular geometries and chemical reactions. The Hellmann-Feynman theorem (electrostatic theorem) states that the force acting on the nucleus A is the vector sum of the repulsive electrostatic forces due to the other nuclei B in the molecule and the attractive forces exerted on the nucleus A from the negatively charged electron cloud of the molecule.
The ESF model predicts the molecular geometry and the course of the chemical reaction by considering the vector sum of these forces on the constituent nuclei.
- Electrostatic Force Theory for a Molecule and Interacting Molecules I. Concept and Illustrative Applications, H. Nakatsuji, J. Am. Chem. Soc., 95(2), 345 (1973).
- Force Models for Molecular Geometry, H. Nakatsuji and T. Koga, in The Force Concept in Chemistry, B. M. Deb, Ed. (Van Nostrand Reinhold, New York,1981), Chap.3, pp. 137-217.
- Common Natures of the Electron Cloud of the System Undergoing Change in Nuclear Configuration, H. Nakatsuji, J. Am. Chem. Soc., 96(1), 24 (1974).
- Force in SCF Theories, H. Nakatsuji, K. Kanda, and T. Yonezawa, Chem. Phys. Lett., 75(2), 340 (1980).
- Force in SCF Theories. Second Derivative of Potential Energy, H. Nakatsuji, K. Kanda, and T. Yonezawa, J. Chem. Phys., 77, 1961 (1982).
Electronic structure and reactivity of metal-carbon and metal-silicon multiple bonds
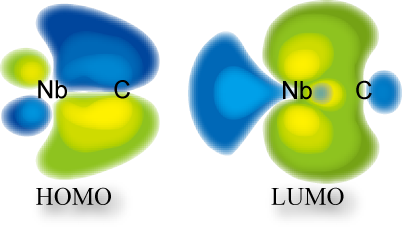
The multiple bond between metal and carbon is of great interest as a typical bonding mode in organometallic chemistry. The formation/breaking of these bonds is a key step in many organometallic reactions. There are two types of metal carbene complexes: Fisher- and Schrock-types. The reactivity of these complexes are different.
In early 1980's, Nakatsuji and co-worker performed SCF-MO calculations. The reactivity of these metal-carbon multiple bonds are understood in a unified form on the basis of the frontier orbital theory. They would never be explained by the charge-controlled mechanism.
- Ab Initio Electronic Structures and Reactivities of Metal Carbene Complexes: Fischer-Type Compounds, (CO)5Cr=CH(OH) and (CO)4Fe=CH(OH), H. Nakatsuji, J. Ushio, S. Han, and T. Yonezawa, J. Am. Chem. Soc., 105, 426 (1983).
- Electronic Structures and Reactivities of Metal-Carbon Multiple Bonds; Schrock-Type Metal-Carbene Complex and Metal-Carbyne Complex, J. Ushio, H. Nakatsuji, and T. Yonezawa, J. Am. Chem. Soc., 106, 5892 (1984).
- Does a Silylene-Metal Complex Exist?, H. Nakatsuji, J. Ushio, and T. Yonezawa, J. Organometal. Chem., 258, C1-C4 (1983).